Hemochromatóza – příčiny
Hemochromatóza – dědičné polysystémové onemocnění provázené aktivním vstřebáváním železa v trávicím traktu a jeho následnou akumulací ve vnitřních orgánech (srdce, slinivka, játra, klouby, hypofýza). Klinický obraz hemochromatózy je charakterizován bronzovou pigmentací kůže a sliznic, rozvojem jaterní cirhózy, diabetes mellitus, kardiomyopatie, artralgie, sexuální dysfunkce aj. Diagnóza hemochromatózy je potvrzena stanovením zvýšeného vylučování železa močí. , vysoký obsah železa v krevním séru a jaterních biopsiích, stejně jako s pomocí rentgenu, ultrazvuku, MRI vnitřních orgánů. Léčba pacientů s hemochromatózou je založena na dietě, podávání deferoxaminu, prokrvení, plazmaferéze, hemosorpci a symptomatické terapii. V případě potřeby se řeší otázka transplantace jater a artroplastiky.
- Příčiny hemochromatózy
- Příznaky hemochromatózy
- Diagnóza hemochromatózy
- Léčba hemochromatózy
- Prognóza a prevence
- Ceny za ošetření
Přehled
Hemochromatóza (bronzový diabetes, pigmentová cirhóza) je geneticky podmíněná porucha metabolismu železa, vedoucí k ukládání pigmentů obsahujících železo ve tkáních a orgánech a rozvoji mnohočetného orgánového selhání. Onemocnění provázené charakteristickým komplexem příznaků (kožní pigmentace, jaterní cirhóza a diabetes mellitus) bylo popsáno v roce 1871 a v roce 1889 dostalo název hemochromatóza pro charakteristické zbarvení kůže a vnitřních orgánů. Incidence hereditární hemochromatózy v populaci je 1,5-3 případy na 1000 obyvatel. Muži trpí hemochromatózou 2-3krát častěji než ženy. Průměrný věk vývoje patologie je 40-60 let. Vzhledem k polysystémové povaze léze se studiem hemochromatózy zabývají různé klinické obory: gastroenterologie, kardiologie, endokrinologie, revmatologie atd.
Po etiologickém aspektu se rozlišuje primární (hereditární) a sekundární hemochromatóza. Primární hemochromatóza je spojena s poruchou enzymových systémů, což vede k ukládání železa ve vnitřních orgánech. V závislosti na genovém defektu a klinickém obrazu existují 4 formy hereditární hemochromatózy:
- I – klasický autozomálně recesivní typ spojený s HFE (více než 95 % případů)
- II – juvenilní typ
- III – dědičný typ neasociovaný s HFE (mutace v transferinovém receptoru typu 2)
- IV – autosomálně dominantní typ.
Sekundární hemochromatóza (generalizovaná hemosideróza) vzniká v důsledku získaného deficitu enzymových systémů zapojených do metabolismu železa a je často spojena s dalšími onemocněními, v souvislosti s nimiž se rozlišují tyto varianty: potransfuzní, alimentární, metabolická, smíšená a novorozenecká .
V klinickém průběhu prochází hemochromatóza 3 stádii: I – bez přetížení železem; II – s přetížením železem, ale bez klinických příznaků; III – s rozvojem klinických projevů.
Příčiny hemochromatózy
Primární hereditární hemochromatóza je onemocnění s autozomálně recesivním typem přenosu. Je založen na mutacích v genu HFE, který se nachází na krátkém raménku 6. chromozomu. Defekt v genu HFE vede k narušení transferinem zprostředkovaného vychytávání železa buňkami duodena, což má za následek vytvoření falešného signálu o nedostatku železa v těle. To zase podporuje zvýšenou syntézu proteinu vázajícího železo DCT-12 enterocyty a zvýšenou absorpci železa ve střevě (při normálním příjmu mikroelementu s potravou). Následně dochází k nadměrnému ukládání pigmentu hemosiderinu obsahujícího železo v mnoha vnitřních orgánech, odumírání jejich funkčně aktivních prvků s rozvojem sklerotických procesů. Při hemochromatóze se ročně nahromadí v lidském těle 1-0,5 g železa a příznaky onemocnění se projeví, když celková hladina železa dosáhne 1,0 g (někdy 20-40 g i více).
Sekundární hemochromatóza vzniká v důsledku nadměrného exogenního příjmu železa do těla. Tento stav může nastat při častých opakovaných krevních transfuzích, nekontrolovaném příjmu preparátů železa, talasémii, některých typech chudokrevnosti, kožní porfyrii, alkoholické cirhóze jater, chronické virové hepatitidě B a C, zhoubných novotvarech a při dodržování nízkoproteinové diety.
Příznaky hemochromatózy
Ke klinické manifestaci hereditární hemochromatózy dochází v dospělosti, kdy celkový obsah železa v těle dosahuje kritických hodnot (20-40 g). V závislosti na převažujících syndromech se rozlišují hepatopatické (hemochromatóza jater), kardiopatické (hemochromatóza srdce) a endokrinologické formy onemocnění.
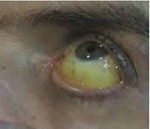
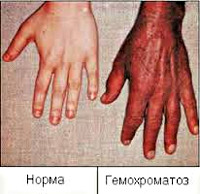
Nemoc se vyvíjí postupně; V počáteční fázi převažují nespecifické stížnosti na zvýšenou únavu, slabost, úbytek na váze a snížení libida. V této fázi mohou pacienty obtěžovat bolesti v pravém hypochondriu, suchá kůže, artralgie způsobená chondrokalcinózou velkých kloubů. V pokročilém stadiu hemochromatózy se tvoří klasický symptomový komplex reprezentovaný kožní pigmentací (bronzová kůže), jaterní cirhózou, diabetes mellitus, kardiomyopatií a hypogonadismem.
Obvykle je nejčasnějším příznakem hemochromatózy objevení se specifického zbarvení kůže a sliznic, vyjádřeného především na obličeji, krku, horních končetinách, v podpaží a zevních genitáliích a kožních jizvách. Intenzita pigmentace závisí na délce onemocnění a pohybuje se od světle šedé (kouřové) až po bronzově hnědou. Mezi charakteristické znaky patří vypadávání vlasů na hlavě a těle a konkávní (lžícovitá) deformace nehtů. Jsou pozorovány artropatie metakarpofalangeálních, někdy kolenních, kyčelních a loketních kloubů s následným rozvojem jejich tuhosti.
Téměř všichni pacienti mají zvětšená játra, splenomegalii a jaterní cirhózu. Dysfunkce slinivky břišní vede k rozvoji inzulin-dependentního diabetes mellitus. V důsledku poškození hypofýzy při hemochromatóze trpí sexuální funkce: u mužů se rozvine testikulární atrofie, impotence a gynekomastie; u žen – amenorea a neplodnost. Srdeční hemochromatóza je charakterizována kardiomyopatií a jejími komplikacemi – arytmie, chronické srdeční selhání, infarkt myokardu.
V terminálním stadiu hemochromatózy se rozvíjí portální hypertenze, ascites a kachexie. Smrt pacientů obvykle nastává v důsledku krvácení z jícnových varixů, selhání jater, akutního srdečního selhání, diabetického kómatu, aseptické peritonitidy a sepse. Hemochromatóza významně zvyšuje riziko vzniku rakoviny jater (hepatocelulárního karcinomu).
Diagnóza hemochromatózy
V závislosti na převažujících příznacích mohou pacienti s hemochromatózou vyhledat pomoc u různých odborníků: gastroenterolog, kardiolog, endokrinolog, gynekolog, urolog, revmatolog, dermatolog. Mezitím je diagnóza onemocnění stejná pro různé klinické varianty hemochromatózy. Po posouzení klinických příznaků je pacientům předepsán soubor laboratorních a instrumentálních studií k zajištění platnosti diagnózy.
Laboratorní kritéria pro hemochromatózu zahrnují významné zvýšení hladiny železa, feritinu a transferinu v krevním séru, zvýšení vylučování železa močí a snížení celkové vazebné kapacity pro železo v krevním séru. Diagnóza je potvrzena punkční biopsií jater nebo kůže, při které je ve vzorcích detekována depozice hemosiderinu. Dědičná povaha hemochromatózy je stanovena jako výsledek molekulárně genetické diagnostiky.
Pro posouzení závažnosti poškození vnitřních orgánů a prognózy onemocnění se vyšetřují jaterní testy, hladina glukózy v krvi a moči, glykosylovaný hemoglobin atd. Laboratorní diagnostika hemochromatózy je doplněna o instrumentální studie: RTG of kloubů, EKG, echokardiografie, ultrazvuk břišních orgánů, MRI jater atd.
Léčba hemochromatózy
Hlavním cílem terapie je odstranit přebytečné železo z těla a zabránit rozvoji komplikací. Pacientům s hemochromatózou je předepsána dieta, která zahrnuje omezení potravin s vysokým obsahem železa (jablka, maso, játra, pohanka, špenát atd.) a lehce stravitelné sacharidy. Je zakázáno užívat multivitaminy, kyselinu askorbovou, doplňky stravy obsahující železo a alkohol. K odstranění přebytečného železa z těla se používá prokrvení pod kontrolou hladiny hemoglobinu, krevního hematokritu a feritinu. Ke stejnému účelu lze použít mimotělní metody hemokorekce – plazmaferéza, hemosorpce, cytaferéza.
Patogenetická medikamentózní terapie hemochromatózy je založena na intramuskulární nebo intravenózní aplikaci deferoxaminu pacientovi, který váže ionty Fe3+. Současně se provádí symptomatická léčba jaterní cirhózy, srdečního selhání, diabetes mellitus a hypogonadismu. V případě těžké artropatie se zjišťují indikace k artroplastice (endoprotetika postižených kloubů). U pacientů s cirhózou se řeší otázka transplantace jater.
Prognóza a prevence
I přes progresivní průběh onemocnění může včasná terapie prodloužit život pacientů s hemochromatózou na několik desetiletí. Bez léčby průměrná délka života pacientů po diagnóze patologie nepřesahuje 4-5 let. Přítomnost komplikací hemochromatózy (zejména jaterní cirhóza a městnavé srdeční selhání) je prognosticky nepříznivým znakem.
U dědičné hemochromatózy spočívá prevence v rodinném screeningu, včasném odhalení a zahájení léčby onemocnění. Vyvážená strava, sledování předepisování a příjmu přípravků železa, krevní transfuze, zdržování se alkoholu a sledování pacientů s onemocněním jater a krevního systému pomáhá předcházet rozvoji sekundární hemochromatózy.