Projev a léčba spondyloepifýzové dysplazie

Základem spondyloepifýzové dysplazie je vývojová vada kloubní chrupavky.
Spondyloepifýzová dysplazie, vrozená forma OMIM 183900, pozdní forma – OMIM 313400 (OMIM je číslo v mezinárodním katalogu dědičných chorob), stručný popis klinického obrazu, kombinovaná patologie vnitřních orgánů, změny inteligence, poměru pohlaví, frekvence v populaci, typ dědictví).
Spondyloepifýzová dysplazie je heterogenní skupina systémových onemocnění kostí s převládajícím poškozením obratlových těl a epifýz tubulárních kostí s tvorbou neúměrně nízkého vzrůstu v důsledku zkrácení trupu, deformací hrudníku a rozvojem degenerativních změn v kloubech.
Etiologie a patogeneze onemocnění nejsou známy. Vrozená forma je autozomálně dominantní dědičnost (poruchy v genu kolagenu typu II COL2A1), pozdní forma je X-vázaná recesivní (mutace v SEDL genu).
Spondyloepifýzová dysplazie – skupina dědičných onemocnění skeletu, která se liší od spondyloepimetafyzárních dysplazií absencí poškození metafýz dlouhých tubulárních kostí
• Vrozená spondyloepifýzová dysplazie (#183900, gen kolagenu COL2A1 [120140]).
Klinicky: vrozený nanismus s krátkým trupem, normocefalie, plochý obličej, myopie, odchlípení sítnice, rozštěp patra, platyspondylie, krátký krk, subluxace krčních obratlů, hypoplazie odontoidního výběžku, kyfóza, skolióza, lumbální lordóza, cervikální myelopatie retardace, sudovitý hrudník, senzorineurální nedoslýchavost, hypoplazie břišních svalů, břišní a tříselné kýly, nedostatečná osifikace stydkých kostí, distální epifýzy femuru a proximální tibie, talu a patní kosti, zploštění těl obratlů
• Dysplazie spondyloepifyzární Maroteaux (184095): platyspondylie, normální inteligence, zkrácení končetin, deformace nohou typu klepání, abnormální tvar pánevního vchodu
• Spondyloepifýzová dysplazie s retinální dystrofií (183850)
• Spondyloepifýzová dysplazie, myopie a senzorineurální ztráta sluchu (184000 XNUMX), možná alelická se Sticklerovým syndromem.
• Schimkeho spondyloepifýzová dysplazie (*242900)
• Spondyloepifýzová dysplazie, typ Irapa (*271650), je běžná u indiánů Irapa z Venezuely a Mexika. Klinické: zkrácená páteř, platyspondylie, krátké záprstní a metatarzální kosti, abnormální proximální femorální a distální epifýzy humeru
• Spondyloepifýzová dysplazie s atlantoaxiální nestabilitou (600561)
• Pseudoachondroplastická spondyloepifýzová dysplazie (3 typy: 177150; 264150; #177170 [19p13.1, COMP gen pro oligomerní protein matrix chrupavky]) je jednou z nejčastějších kostních dysplazií. Pacienti se při narození zdají normální a retardace růstu je zřídka rozpoznána až do druhého roku života nebo později. Na rozdíl od achondroplazie jsou hlava a obličej normální. Prsty jsou krátké, ale nemají tvar trojzubce typický pro achondroplazii. Deformace dolních končetin jsou různé, je zaznamenána slabost vazů.
Klinické: nanismus s krátkými končetinami, rozpoznatelný v dětství; bederní lordóza, kyfóza, skolióza, luxace v atlantoaxiálním kloubu, brachydaktylie, ulnární deviace zápěstí, omezení extenze v loketních a kyčelních kloubech, ochablost vazů, deformace kolena, chronická myelopatie krční míchy, platyspondylie, deformace obratlových těl, zkrácení tubulárních kostí, rozšíření metafýz, abnormální epifýzy
• Spondyloepifýzová dysplazie, pozdně dominantní (*184100): nanismus se zkráceným trupem rozpoznatelným v dětství, široký obličej, platyspondylie, krátký krk, subluxace krčních obratlů, hypoplazie odontoidního výběžku, kyfoskolióza, bederní lordóza, sudovitý hrudník, patologie hlavice stehenní kosti s degenerativními změnami
• Spondyloepifýzová dysplazie pozdní s charakteristickou facie (600093): mikrocefalie, opožděný vývoj, široký kořen a špička nosu, krátké široké filtrum, tlusté rty, progresivní zúžení meziobratlových prostor, zploštělé genikulární epifýzy
• Pozdní spondyloepifýzová dysplazie s progresivní artropatií (*208230, 6q, gen PPAC). Synonymum: progresivní pseudorevmatoidní artropatie. Klinicky: artropatie, progresivní ranní ztuhlost, otoky kloubů prstů; histologicky: normální synoviální membrána, věk nástupu cca 3 roky, snížená pohyblivost krční páteře, zploštělá těla obratlů, osifikační defekty, rozšířené proximální a střední články prstů. Laboratoř: normální ESR, negativní revmatoidní testy, kostní dysplazie, abnormální acetabulum, malý vzrůst v dospělosti (140–150 cm)
• Spondyloepifýzová dysplazie, pozdní (*313400): vrozený nanismus s krátkými končetinami, normální tvar lebky, plochý obličej, krátký krk, platyspondylie, subluxace krčních obratlů, hypoplazie odontoidního výběžku, kyfoskolióza, degenerativní lordóza hrudníku, bederní arthritida, barel kyčelních kloubů, diagnózu nelze instalovat dříve než ve věku 4-6 let
• Spondyloepifýzová dysplazie, pozdně recesivní (*271600)
• Pozdní spondyloepifýzová dysplazie s mentální retardací (271620).
Klinicky: lehká až středně těžká mentální retardace, jazykovité bederní obratle, platyspondylie, rozšíření kyčelní kosti, deformita acetabula se subluxací kyčle a varózní deformita v kloubu, tenké krčky femuru. MKN-10. Q77.7 Spondyloepifýzová dysplazie.
<strong>Klasifikace</strong>
- 1. Vrozená spondyloepimetafyzární dysplazie.
- 2. Pozdní spondyloepimetafyzární dysplazie (Kozlovského typ).
- 3. Metatropní dysplazie.
- 4. Parastomální dysplazie.
- 5. Diggvi-Melchior-Clausenova nemoc.
<strong>Klinický obraz</strong>
První klinické příznaky se objevují především poté, co se dítě poprvé postaví (tj. zaujme vertikální polohu); Obvykle jsou příznaky onemocnění nejčastěji pozorovány ve věku 5-6 let.
U těžkých forem onemocnění se někdy v prvním roce života objeví symptom jako omezená abdukce v kyčelních kloubech.
Děti jsou zakrnělé.
Ve stoji je bederní lordóza, tělo je nakloněno dozadu, žaludek vyčnívá dopředu. Chůze jako kachna. U větších dětí se trup zkracuje a končetiny se kvůli tomu zdají dlouhé.
Nápadná je slabost svalů dolních končetin. Pacienti často vykazují změny v kardiovaskulárním systému, jako je kardiopatie, zvětšená játra a slezina, tříselné kýly a divergence přímých břišních svalů. Všechny tyto změny ukazují na systémové poškození pojivové tkáně. Stížnosti na rychlou únavu, bolesti v dolní části zad a dolních končetin.
<strong>diagnostika</strong>
Radiologicky jsou změněny epifýzy všech tubulárních kostí, ale nejdramatičtější změny jsou pozorovány na stehenních a tibiových kostech. V kyčelních kloubech vzniká dojem vrozené luxace kyčlí.
Jak dítě roste, objevuje se nepoměr v růstu acetabula a proximálního femuru: acetabulum je vytvořeno správně, ale proximální konec výrazně zaostává ve vývoji.
Všechna těla obratlů jsou zploštělá, více v hrudní oblasti. Předozadní a laterální rozměry obratlových těl jsou normální, koncové ploténky jsou nerovnoměrné. Jejich obrysy jsou zvlněné a strukturované. Meziobratlové prostory jsou zúžené. S věkem je zřetelně patrné ztenčení obratlových oblouků v bederní oblasti.
Dochází ke zpoždění ve výskytu osifikačních jader v kostech zápěstí a tarzu, stejně jako v hlavách záprstních a metatarzálních kostí.
Po 5 letech věku jsou zápěstní kůstky, hlavičky záprstních kůstek a distální epifýzy kostí předloktí zploštělé, ale průřez se nemění, někdy je dokonce širší než epifyzární ploténka a epifýza přečnívá jako houba. V kolenních kloubech dochází k vyhlazení interkondylické eminence tibie a rozšíření interkondylické jamky femuru.
<strong>Diferenciální diagnostika</strong>
Diferenciální diagnostika je obtížná a provádí se u následujících onemocnění:
<strong>Léčba</strong>
Pro dosažení nejlepších výsledků léčba nekončí na operačním stole, ale vyžaduje komplexní individuální pooperační péči. rehabilitace, zahrnující: drogovou rehabilitaci, technické rehabilitační vybavení (ortézy, ortopedická obuv, vertikalizátory od jednoduchých chodítek po systém Locomat), školení ve specializovaných ústavech, lázeňskou rehabilitaci.
Metody chirurgická korekce u této skupiny pacientů jsou stále jedinými účinnými metodami léčby muskuloskeletálních patologií pro zlepšení kvality života a usnadnění sociální adaptace pacientů.
Dědičně podmíněné dysplastické léze muskuloskeletálního systému u dětí, které často vyžadují etapovou a traumatickou chirurgickou léčbu, se přednostně provádějí v specializovaná lékařská centra, kteří mají významné praktické a organizační zkušenosti (intervence, ortotika, funkční rehabilitace) s léčbou této patologie, což zvyšuje efektivitu léčby a snižuje počet komplikací.
Genová terapie dědičná onemocnění s poškozením pohybového aparátu by se v budoucnu měla stát nejúčinnější metodou terapie, v současnosti však zatím nepřesáhla rámec klinických experimentů a je extrémně nákladná.
Prognóza do života je příznivá. Funkční prognóza: časná artróza.
Materiály zkontroloval traumatolog-ortoped: Leontiev V.S.
Byla tato informace užitečná? Pak sdílejte se svými přáteli

Knistova dysplazie – jedna z forem nanismu způsobená genetickými poruchami vedoucími ke vzniku defektních forem jedné z odrůd hlavního proteinu pojivových tkání – kolagenu. Příznaky tohoto stavu lze rozpoznat již při narození – pacienti mají relativní zkrácení končetin a zhoršenou pohyblivost kloubů, někdy je zaznamenán i rozštěp patra. Později je obraz onemocnění doplněn o další projevy nanismu. Diagnostika Knistovy dysplazie je stanovena na základě údajů o stavu pacienta, rentgenového vyšetření skeletu a geneticko-molekulární analýzy. Neexistuje žádná specifická léčba onemocnění, používá se symptomatická a podpůrná terapie.
- Příčiny Knistovy dysplazie
- Příznaky Knistovy dysplazie
- Diagnóza Knistovy dysplazie
- Léčba a prevence Knist dysplazie
- Prognóza Knistovy dysplazie
Přehled
Knistova dysplazie (metatropická dysplazie typu 2) je poměrně vzácné genetické onemocnění charakterizované poruchou tvorby jednotlivých kosterních elementů s rozvojem nanismu a dalších patologií. Tento stav byl poprvé popsán a definován jako dědičná patologie v roce 1952 W. Knistem, od té doby nese tento typ skeletální dysplazie jeho jméno. Vzhledem k nízké prevalenci Kniestovy dysplazie v populaci není zatím možné spolehlivě určit výskyt tohoto stavu. Problém je dále komplikován skutečností, že existuje mnoho velmi podobných syndromů způsobených defekty ve stejném genu (Col2A1). Donedávna se věřilo, že jediným mechanismem dědičnosti Knistovy dysplazie je autozomálně dominantní, ale v posledních letech se objevily náznaky existence autozomálně recesivních forem patologie. To může být způsobeno neúplnou penetrací defektní formy genu Col2A1. Rozdělení Knistovy dysplazie podle pohlaví nemá žádné zvláštní rysy, onemocnění postihuje stejně často chlapce i dívky.

Příčiny Knistovy dysplazie
Hlavní příčinou Knistovy dysplazie jsou mutace v genu Col2A1, který se nachází na 12. chromozomu. Gen kóduje sekvenci jednoho z nejběžnějších proteinů pojivové tkáně – kolagenu typu 2 podtřídy alfa-1, který je široce zastoupen ve strukturách kostí a chrupavek v těle. Kromě Kniestovy dysplazie mohou defekty ve struktuře Col2A1 způsobit tak dobře známé genetické stavy, jako je achondrogeneze typu 2, Legg-Calve-Perthesova choroba, vrozená spondyloepifýzová dysplazie a řada dalších skeletálních anomálií. Důvody, proč některé syndromy vznikají, sahají až do stupně narušení kolagenové struktury vyjádřené defektním genem – čím více je poškozen, tím výraznější budou projevy dysplazie.
V naprosté většině případů se mutace v genu Col2A1, které vedou ke vzniku Kniestovy dysplazie, dědí autozomálně dominantním způsobem. Zprávy některých genetiků o objevu recesivních forem této patologie byly vědeckou komunitou vnímány s pochybami – specialisté předložili předpoklady o možné neúplné penetraci defektního genu, která by mohla být považována za jeho recesivitu. Následně se však objevily závažnější důkazy pro existenci autozomálně recesivních forem Kniestovy dysplazie.
Patologické změny v těle pacientů trpících tímto onemocněním se neomezují pouze na kosterní anomálie – defektní kolagenová struktura může způsobit zhoršenou pohyblivost kloubů, svalovou slabost a poruchu zraku. Někteří pacienti s Knistovou dysplazií také pociťují senzorineurální hluchotu, ale mechanismus rozvoje této anomálie u tohoto onemocnění není znám.
Příznaky Knistovy dysplazie
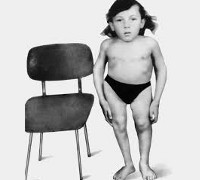
Řadu projevů Knistovy choroby lze registrovat již při narození dítěte, další příznaky se zjišťují v prvních letech života. Mezi nejranější příznaky onemocnění lze u novorozence vyzdvihnout kombinaci kulatého plochého obličeje a velkých a vypouklých očí. V některých případech se uvedené poruchy kombinují s rozštěpem patra. Trup pacientů s Knistovou dysplazií je zkrácen, relativní velikosti končetin jsou zmenšeny a v oblasti kloubů jsou pozorovány ztluštění. Dítě vykazuje svalovou slabost, někdy je zjištěna krátkozrakost a hluchota. Pacient výrazně zaostává za svými vrstevníky ve fyzickém vývoji. Později se u Knistovy dysplazie vyvine zakřivení páteře, na hrudní úrovni se objeví kyfóza a na bederní úrovni výrazná lordóza. V některých případech mohou tyto procesy způsobit dýchací potíže a srdeční problémy, které mohou být smrtelné.
V důsledku výše popsaných kosterních anomálií se u pacientů s Knistovou dysplazií vyvine široký a plochý hrudník, často s prohlubní ve středu. Kontraktury kyčelních, kolenních a loketních kloubů a relativní prodloužení prstů jsou zjištěny poměrně brzy. Někdy se objevuje alopecie. Děti s Knistovou dysplazií začínají mluvit mnohem později než zdraví vrstevníci, ale nejčastěji je to spojeno s poruchami struktury a inervace prvků patra a artikulačního aparátu – intelekt pacientů s tímto onemocněním je téměř vždy zachován. Nedostatečný vývoj řeči může také naznačovat, že pacient je hluchý, také způsobený dědičným onemocněním. Polovina pacientů s Knistovou dysplazií má různé zrakové vady: krátkozrakost, odchlípení sítnice, dislokaci čočky a řadu dalších. Výška dospělého s touto patologií je asi 110-115 centimetrů.
Diagnóza Knistovy dysplazie
Diagnostika Knistovy dysplazie je založena na údajích z fyzikálního vyšetření pacienta, studia jeho rodinné anamnézy, rentgenových a molekulárně genetických studií. Při vyšetření novorozence se odhalí charakteristický vzhled (plochý kulatý obličej, sedlovitý nos, vypouklé oči), zkrácené tělo a končetiny, ztluštění v oblasti kloubu. Jak pacient roste, k těmto projevům Kniestovy dysplazie se připojují zakřivení páteře (hrudní kyfóza a bederní lordóza), kloubní kontraktury, PEC a další anomálie skeletu. U tohoto typu kostní dysplazie mohou mít končetiny různé velikosti. Opožděný vývoj řeči může být spojen jak s vadami artikulačního aparátu, tak se senzorineurální hluchotou pacienta.
Největší množství informací důležitých pro diagnostiku Knistovy dysplazie poskytují radiologické vyšetřovací metody. Snímky páteře ukazují oploštění jednotlivých obratlů (platyspondyly). Dlouhé trubkovité kosti končetin jsou zkrácené, jejich metafýzy jsou často rozšířeny a epifýzy jsou zploštělé. Charakteristickým příznakem Kniestovy dysplazie je rozšíření konců krátkých tubulárních kostí (například falangy prstů). Téměř vždy jsou nalezeny známky porušení metaepifyzární osifikace – osifikační jádra (například v metafýzách stehenních kostí) se objevují mnohem později než obvykle nebo se vůbec nevyvíjejí.
Oftalmologické vyšetření pacienta s Kniestovou dysplazií může odhalit krátkozrakost (myopii), která je časem někdy komplikována odchlípením sítnice a slepotou. Mezi takovými pacienty je zvýšený výskyt pupeční a tříselné kýly. Molekulárně genetická diagnostika Kniestovy dysplazie zahrnuje přímé automatizované sekvenování sekvence genu Col2A1 k identifikaci mutací. Pro vyloučení spíše vzácných autozomálně recesivních forem onemocnění se obdobná analýza provádí u rodičů a příbuzných pacienta, aby se zjistilo, zda jsou nositeli patologického genu. Diferenciální diagnostika Knistovy dysplazie se provádí s mukopolysacharidózou typu 4 a dědičnými syndromy způsobenými defekty genu Col2A1.
Léčba a prevence Knist dysplazie
Pro Knistovu dysplazii neexistuje žádná specifická terapie. Symptomatická léčba může zahrnovat chirurgickou korekci rozštěpu patra, prováděnou v prvních měsících života dítěte. Kromě toho může Kniestova dysplazie vyžadovat chirurgické zákroky ke zvýšení pohyblivosti kloubů v případech těžkých kontraktur. Krátkozrakost spojená s tímto onemocněním je poměrně obtížně léčitelná, ale pro včasné odhalení a odstranění odchlípení sítnice je nutné pravidelné sledování oftalmologem. Podpůrná terapie Knistovy dysplazie zahrnuje užívání vitamínových a minerálních komplexů, dietu se zvýšeným obsahem bílkovin a snížení zátěže kloubů. Vzhledem k převážně dominantnímu charakteru onemocnění nebyla vyvinuta preventivní opatření.
Prognóza Knistovy dysplazie
Knist dysplazie je dosti závažnou variantou dědičné anomálie skeletu a nanismu a často vede k invaliditě pacienta v raném věku. V závažných případech onemocnění může zakřivení páteře a deformace hrudníku vyvolat respirační nebo kardiovaskulární selhání. Celkově je prognóza Knistovy dysplazie špatná. Mnoho pacientů se sice dožívá dospělosti, ale jejich délka života se oproti zdravým lidem výrazně zkracuje. Smrt nejčastěji nastává v důsledku sekundárních poruch fungování vnitřních orgánů způsobených závažnými kosterními abnormalitami a vývojovými vadami.